Using sample representation methods#
In this tutorial, we will demostrate how to use patpy to build sample representation from single-cell transcriptomics data, and how to compare representations from different methods
Install the patpy package#
This package contains interface to sample representation methods as well as some handy analysis functions.
!pip install git+https://github.com/lueckenlab/patpy.git@main
Import packages#
import ehrapy as ep
import pandas as pd
import scanpy as sc
import patpy
import seaborn as sns
from plottable import ColumnDefinition, Table
from plottable.cmap import normed_cmap
from plottable.plots import bar
import matplotlib
import matplotlib.pyplot as plt
from matplotlib.colors import LinearSegmentedColormap
patpy.__version__
'0.9.0'
Read the data#
Here, we use COMBAT dataset. This dataset contains 783k cells from 140 COVID-19 patients and healthy donors.
ADATA_PATH = "/Users/vladimir.shitov/Documents/programming/pat_rep_benchmark/data/combat/combat_processed.h5ad"
adata = sc.read_h5ad(ADATA_PATH)
adata
AnnData object with n_obs × n_vars = 783677 × 3000
obs: 'Annotation_cluster_id', 'Annotation_cluster_name', 'Annotation_minor_subset', 'Annotation_major_subset', 'Annotation_cell_type', 'GEX_region', 'QC_ngenes', 'QC_total_UMI', 'QC_pct_mitochondrial', 'QC_scrub_doublet_scores', 'TCR_chain_composition', 'TCR_clone_ID', 'TCR_clone_count', 'TCR_clone_proportion', 'TCR_contains_unproductive', 'TCR_doublet', 'TCR_chain_TRA', 'TCR_v_gene_TRA', 'TCR_d_gene_TRA', 'TCR_j_gene_TRA', 'TCR_c_gene_TRA', 'TCR_productive_TRA', 'TCR_cdr3_TRA', 'TCR_umis_TRA', 'TCR_chain_TRA2', 'TCR_v_gene_TRA2', 'TCR_d_gene_TRA2', 'TCR_j_gene_TRA2', 'TCR_c_gene_TRA2', 'TCR_productive_TRA2', 'TCR_cdr3_TRA2', 'TCR_umis_TRA2', 'TCR_chain_TRB', 'TCR_v_gene_TRB', 'TCR_d_gene_TRB', 'TCR_j_gene_TRB', 'TCR_c_gene_TRB', 'TCR_productive_TRB', 'TCR_chain_TRB2', 'TCR_v_gene_TRB2', 'TCR_d_gene_TRB2', 'TCR_j_gene_TRB2', 'TCR_c_gene_TRB2', 'TCR_productive_TRB2', 'TCR_cdr3_TRB2', 'TCR_umis_TRB2', 'BCR_umis_HC', 'BCR_contig_qc_HC', 'BCR_functionality_HC', 'BCR_v_call_HC', 'BCR_v_score_HC', 'BCR_j_call_HC', 'BCR_j_score_HC', 'BCR_junction_aa_HC', 'BCR_total_mut_HC', 'BCR_s_mut_HC', 'BCR_r_mut_HC', 'BCR_c_gene_HC', 'BCR_clone_per_replicate_HC', 'BCR_clone_global_HC', 'BCR_clonal_abundance_HC', 'BCR_locus_LC', 'BCR_umis_LC', 'BCR_contig_qc_LC', 'BCR_functionality_LC', 'BCR_v_call_LC', 'BCR_v_score_LC', 'BCR_j_call_LC', 'BCR_j_score_LC', 'BCR_junction_aa_LC', 'BCR_total_mut_LC', 'BCR_s_mut_LC', 'BCR_r_mut_LC', 'BCR_c_gene_LC', 'COMBAT_ID', 'scRNASeq_sample_ID', 'COMBAT_participant_timepoint_ID', 'Source', 'Age', 'Sex', 'Race', 'BMI', 'Hospitalstay', 'Death28', 'Institute', 'PreExistingHeartDisease', 'PreExistingLungDisease', 'PreExistingKidneyDisease', 'PreExistingDiabetes', 'PreExistingHypertension', 'PreExistingImmunocompromised', 'Smoking', 'Symptomatic', 'Requiredvasoactive', 'Respiratorysupport', 'SARSCoV2PCR', 'Outcome', 'TimeSinceOnset', 'Ethnicity', 'Tissue', 'DiseaseClassification', 'Pool_ID', 'Channel_ID', 'ifn_1_score', '_scvi_batch', '_scvi_labels', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_50_genes', 'pct_counts_in_top_100_genes', 'pct_counts_in_top_200_genes', 'pct_counts_in_top_500_genes', 'total_counts_mt', 'log1p_total_counts_mt', 'pct_counts_mt', 'total_counts_ribo', 'log1p_total_counts_ribo', 'pct_counts_ribo'
var: 'gene_ids', 'feature_types', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm', 'highly_variable_nbatches', 'mt', 'ribo', 'n_cells_by_counts', 'mean_counts', 'log1p_mean_counts', 'pct_dropout_by_counts', 'total_counts', 'log1p_total_counts'
uns: 'Institute', 'ObjectCreateDate', 'Source_colors', 'Technology', 'X_scpoli', '_scvi_manager_uuid', '_scvi_uuid', 'genome_annotation_version', 'hvg', 'log1p', 'pca', 'scpoli_distances', 'scpoli_parameters', 'scpoli_samples'
obsm: 'X_pca', 'X_scANVI_batch', 'X_scANVI_sample', 'X_scVI_batch', 'X_scVI_sample', 'X_scpoli', 'X_umap', 'X_umap_source'
varm: 'PCs'
layers: 'X_raw_counts'
Set columns containing sample IDs, cell types and metadata#
sample_id_col = "scRNASeq_sample_ID"
cell_type_key = "cell_type"
samples_metadata_cols = ["Source", "Outcome", "Death28", "Institute", "Pool_ID"]
Currently, there is no such columns as “cell_type” in the data. But cell types are stored in the Annotation_major_subset column. Let’s rename it to cell_type for better readability.
adata.obs.rename(columns={"Annotation_major_subset": cell_type_key}, inplace=True)
Store metadata and calculate QC metrics#
metadata = adata.obs[samples_metadata_cols + [sample_id_col]].drop_duplicates()
metadata.set_index(sample_id_col, inplace=True)
metadata
| Source | Outcome | Death28 | Institute | Pool_ID | |
|---|---|---|---|---|---|
| scRNASeq_sample_ID | |||||
| S00109-Ja001E-PBCa | COVID_SEV | 2.0 | 0 | Oxford | gPlexA |
| S00112-Ja003E-PBCa | COVID_MILD | 5.0 | 0 | Oxford | gPlexA |
| S00005-Ja005E-PBCa | COVID_CRIT | 2.0 | 0 | Oxford | gPlexA |
| S00061-Ja003E-PBCa | COVID_SEV | 4.0 | 0 | Oxford | gPlexA |
| S00056-Ja003E-PBCa | COVID_SEV | 3.0 | 0 | Oxford | gPlexA |
| ... | ... | ... | ... | ... | ... |
| S00065-Ja003E-PBCa | COVID_CRIT | 2.0 | 0 | Oxford | gPlexK |
| S00048-Ja003E-PBCa | COVID_SEV | 4.0 | 0 | Oxford | gPlexK |
| G05112-Ja005E-PBCa | COVID_HCW_MILD | 6.0 | 0 | Oxford | gPlexK |
| N00038-Ja001E-PBGa | Sepsis | NaN | 0 | Oxford | gPlexK |
| U00501-Ua005E-PBUa | Flu | 2.0 | 0 | St_Georges | gPlexK |
138 rows × 5 columns
cell_qc_metadata = patpy.pp.calculate_cell_qc_metrics(
adata, sample_key=sample_id_col, cell_qc_vars=["QC_ngenes", "QC_pct_mitochondrial", "QC_scrub_doublet_scores"]
)
cell_qc_metadata
| median_QC_ngenes | median_QC_pct_mitochondrial | median_QC_scrub_doublet_scores | |
|---|---|---|---|
| scRNASeq_sample_ID | |||
| G05061-Ja005E-PBCa | 1107.0 | 3.011159 | 0.050648 |
| G05064-Ja005E-PBCa | 975.0 | 1.332430 | 0.060894 |
| G05073-Ja005E-PBCa | 1141.0 | 2.422559 | 0.044530 |
| G05077-Ja005E-PBCa | 1125.0 | 2.946723 | 0.048490 |
| G05078-Ja005E-PBCa | 999.0 | 2.825308 | 0.052783 |
| ... | ... | ... | ... |
| U00607-Ua005E-PBUa | 1827.0 | 2.982509 | 0.043323 |
| U00613-Ua005E-PBUa | 1251.5 | 2.053083 | 0.036956 |
| U00617-Ua005E-PBUa | 1410.5 | 3.886215 | 0.057906 |
| U00619-Ua005E-PBUa | 1532.0 | 2.688350 | 0.041823 |
| U00701-Ua005E-PBUa | 1093.5 | 3.714650 | 0.063941 |
138 rows × 3 columns
n_genes_metadata = patpy.pp.calculate_n_cells_per_sample(adata, sample_id_col)
n_genes_metadata
| n_cells | |
|---|---|
| scRNASeq_sample_ID | |
| S00052-Ja005E-PBCa | 13918 |
| H00054-Ha001E-PBGa | 10938 |
| H00067-Ha001E-PBGa | 10781 |
| N00023-Ja001E-PBGa | 10484 |
| H00053-Ha001E-PBGa | 10458 |
| ... | ... |
| U00607-Ua005E-PBUa | 1021 |
| U00613-Ua005E-PBUa | 970 |
| U00701-Ua005E-PBUa | 872 |
| U00601-Ua005E-PBUa | 619 |
| U00504-Ua005E-PBUa | 161 |
138 rows × 1 columns
composition_metadata = patpy.pp.calculate_compositional_metrics(adata, sample_id_col, [cell_type_key], normalize_to=100)
composition_metadata
| cell_type | cell_type_B | cell_type_CD4 | cell_type_CD8 | cell_type_DC | cell_type_DN | cell_type_DP | cell_type_GDT | cell_type_HSC | cell_type_MAIT | cell_type_Mast | cell_type_NK | cell_type_PB | cell_type_PLT | cell_type_RET | cell_type_cMono | cell_type_iNKT | cell_type_ncMono |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| scRNASeq_sample_ID | |||||||||||||||||
| G05061-Ja005E-PBCa | 6.324900 | 33.921438 | 12.366844 | 1.597870 | 0.532623 | 0.499334 | 0.898802 | 0.066578 | 4.677097 | 0.000000 | 18.159121 | 0.316245 | 0.166445 | 0.016644 | 15.812250 | 0.033289 | 4.610519 |
| G05064-Ja005E-PBCa | 3.405158 | 47.147482 | 16.400581 | 1.819806 | 1.228090 | 0.725689 | 2.188233 | 0.022329 | 1.317405 | 0.000000 | 7.457854 | 0.446578 | 0.000000 | 0.000000 | 14.357486 | 0.000000 | 3.483309 |
| G05073-Ja005E-PBCa | 5.194338 | 45.609405 | 16.278791 | 1.487524 | 1.247601 | 0.839731 | 4.654511 | 0.011996 | 2.195298 | 0.011996 | 3.730806 | 0.203935 | 0.047985 | 0.000000 | 13.963532 | 0.083973 | 4.438580 |
| G05077-Ja005E-PBCa | 5.846211 | 29.231056 | 14.596909 | 1.377770 | 0.446844 | 1.340532 | 0.465463 | 0.167567 | 0.800596 | 0.018619 | 22.844908 | 1.079873 | 0.074474 | 0.018619 | 18.004096 | 0.055856 | 3.630609 |
| G05078-Ja005E-PBCa | 1.366381 | 39.000106 | 15.591569 | 2.340854 | 0.762631 | 0.730855 | 2.648025 | 0.211842 | 1.737104 | 0.021184 | 10.666243 | 0.148289 | 0.021184 | 0.000000 | 19.521237 | 0.497829 | 4.734668 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| U00607-Ua005E-PBUa | 3.623898 | 10.577865 | 1.273262 | 4.701273 | 0.097943 | 0.195886 | 0.195886 | 0.685602 | 0.097943 | 0.000000 | 5.876592 | 1.175318 | 1.958864 | 0.097943 | 37.904016 | 0.000000 | 31.537708 |
| U00613-Ua005E-PBUa | 7.835052 | 26.391753 | 16.907216 | 0.721649 | 0.412371 | 0.309278 | 1.649485 | 0.000000 | 0.103093 | 0.000000 | 5.154639 | 1.237113 | 0.206186 | 0.000000 | 37.938144 | 0.000000 | 1.134021 |
| U00617-Ua005E-PBUa | 2.977233 | 41.418564 | 16.462347 | 0.437828 | 0.525394 | 0.262697 | 0.262697 | 0.963222 | 0.087566 | 0.000000 | 7.530648 | 17.513135 | 0.788091 | 0.087566 | 9.719790 | 0.000000 | 0.963222 |
| U00619-Ua005E-PBUa | 3.537618 | 35.077230 | 15.894370 | 0.896861 | 0.697559 | 0.298954 | 2.441455 | 1.145989 | 0.099651 | 0.099651 | 17.588440 | 2.840060 | 4.534131 | 0.000000 | 10.363727 | 0.000000 | 4.484305 |
| U00701-Ua005E-PBUa | 1.032110 | 15.596330 | 39.334862 | 1.261468 | 0.802752 | 0.458716 | 0.573394 | 0.114679 | 0.000000 | 0.000000 | 12.844037 | 0.000000 | 0.344037 | 0.229358 | 24.885321 | 0.000000 | 2.522936 |
138 rows × 17 columns
Merge metadata tables
metadata = pd.concat(
[
metadata,
cell_qc_metadata.loc[metadata.index],
n_genes_metadata.loc[metadata.index],
composition_metadata.loc[metadata.index],
],
axis=1,
)
metadata.shape

Quality control#
To reduce noise in the representations, we need to remove samples with too few cells:
adata = patpy.pp.filter_small_samples(adata, sample_key=sample_id_col, sample_size_threshold=100)
0 samples removed:
If necessary, we can also remove cell types with too few cells in at least one sample:
adata = patpy.pp.filter_small_cell_types(adata, sample_key=sample_id_col, cell_group_key=cell_type_key, cell_type_size_threshold=10)
Some methods require this filtering step prior to building representation. In this notebook, we’ll focus on simpler methods that can be used with only sample filtering.
Run a simple pseudobulk representation#
This method aggregates all cells from each sample into a single vector. It can be interpreted as an average cell from each sample. Any input space can be used specified by layer parameter. For example, to use count data, use layer="X" or layer="raw". We find batch-corrected space to be the best for the representation, so we use layer="X_scVI" below.
pseudobulk = patpy.tl.Pseudobulk(sample_key=sample_id_col, cell_group_key=cell_type_key, layer="X_scVI_batch")
pseudobulk.prepare_anndata(adata)
Calculate a matrix of distances between samples#
distances = pseudobulk.calculate_distance_matrix(force=True)
distances
array([[0. , 2.13826083, 3.26751086, ..., 2.26537604, 3.47556017,
3.50837059],
[2.13826083, 0. , 2.57066436, ..., 2.67332482, 3.12726692,
2.3833747 ],
[3.26751086, 2.57066436, 0. , ..., 3.81414183, 3.50147504,
2.11261287],
...,
[2.26537604, 2.67332482, 3.81414183, ..., 0. , 3.55756191,
4.0558818 ],
[3.47556017, 3.12726692, 3.50147504, ..., 3.55756191, 0. ,
2.70576771],
[3.50837059, 2.3833747 , 2.11261287, ..., 4.0558818 , 2.70576771,
0. ]])
We can also see the pseudobulks for each sample:
pseudobulk.sample_representation
array([[-0.39708972, -0.1610353 , -0.60592145, ..., 0.04138801,
-0.23706943, 0.03939089],
[-0.16564418, 0.15338482, -0.00623349, ..., -0.36909145,
-0.61890358, -0.26146826],
[-0.91906726, -0.34432968, -0.03022187, ..., -1.03792512,
-0.26926878, 0.30427763],
...,
[ 0.07143331, -0.52035475, -0.04083514, ..., -0.01975986,
-0.2641018 , -0.32993519],
[-0.86681354, -0.71001476, 0.40946293, ..., 0.38672951,
0.59467804, 0.03230282],
[-0.98729014, -0.46418488, 0.28515279, ..., -0.19687848,
-0.42034405, 0.18162856]])
We can then visualise sample representation using dimensionality reduction methods:
pseudobulk.plot_embedding(method="UMAP", metadata_cols=samples_metadata_cols);
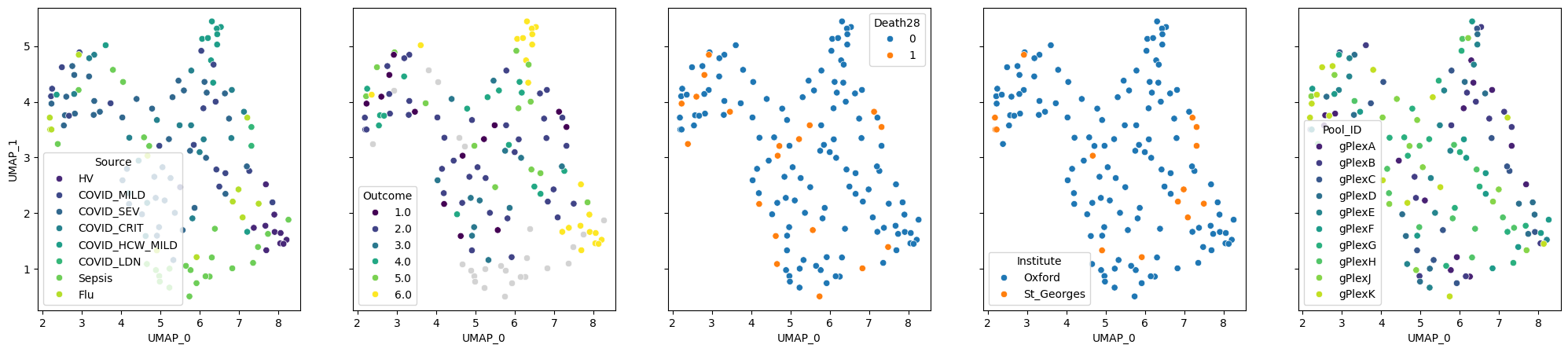
Other dimensionality reduction methods are also available
pseudobulk.plot_embedding(method="TSNE", metadata_cols=samples_metadata_cols);
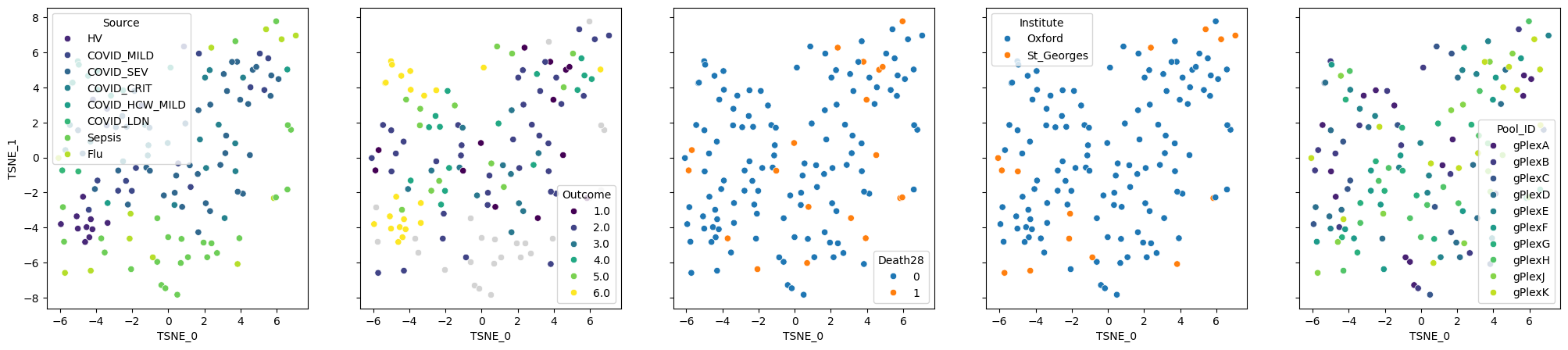
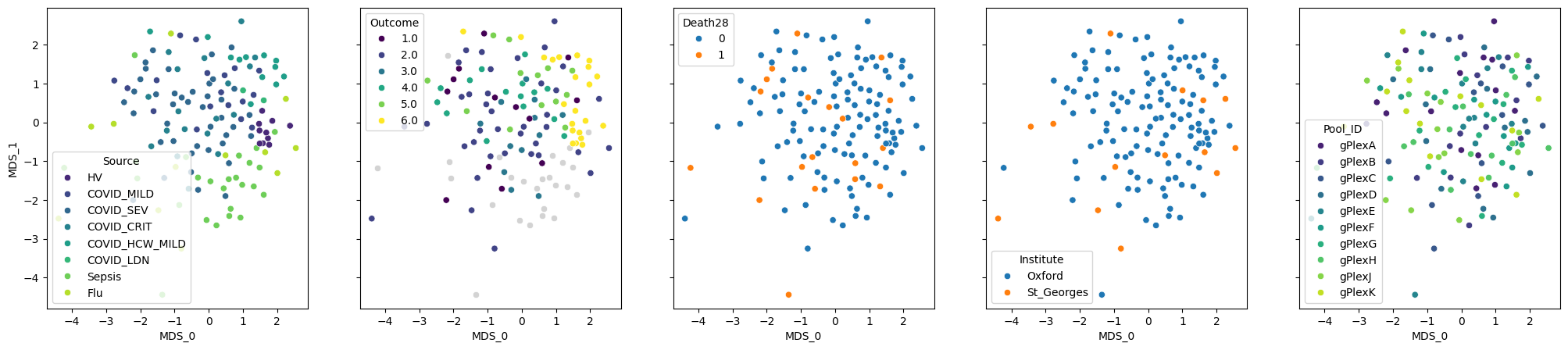
pseudobulk.plot_embedding(method="MDS", metadata_cols=samples_metadata_cols);
Evaluate how well a covariate is represented#
Let’s try to classify “Outcome” based on the nearest neighbors for each sample in the representation:
pseudobulk.evaluate_representation(target="Outcome", method="knn", n_neighbors=5, task="classification")
{'score': 0.13977126662840952,
'metric': 'f1_macro_calibrated',
'n_unique': 6,
'n_observations': 113,
'method': 'knn'}
It doesn’t work too good. Now we can try to solve the ranking problem for the same covariate. It will train regressor and use a different metric for evaluation:
pseudobulk.evaluate_representation(target="Outcome", method="knn", n_neighbors=5, task="ranking")
{'score': 0.6103813851943187,
'metric': 'spearman_r',
'n_unique': 6,
'n_observations': 113,
'method': 'knn'}
Now let’s see how well Pool is represented. This is a technical covariate so we don’t want score to be high:
pseudobulk.evaluate_representation(target="Pool_ID", method="knn", n_neighbors=5, task="classification")
{'score': 0.13579292359838518,
'metric': 'f1_macro_calibrated',
'n_unique': 10,
'n_observations': 138,
'method': 'knn'}
Save the distances to adata to use later
adata.uns["pseudobulk_distances"] = distances
adata.uns["pseudobulk_samples"] = pseudobulk.samples
adata.uns["pseudobulk_UMAP"] = pseudobulk.embeddings["UMAP"]
Run cell type composition representation#
This representation is based on cell type composition differences. It is calculated as a difference between cell type proportions in each sample.
composition = patpy.tl.CellGroupComposition(sample_key=sample_id_col, cell_group_key=cell_type_key, layer="X_scVI")
composition.prepare_anndata(adata)
composition_distances = composition.calculate_distance_matrix(force=True)
composition.plot_embedding(method="UMAP", metadata_cols=samples_metadata_cols)
array([<Axes: xlabel='UMAP_0', ylabel='UMAP_1'>,
<Axes: xlabel='UMAP_0', ylabel='UMAP_1'>,
<Axes: xlabel='UMAP_0', ylabel='UMAP_1'>,
<Axes: xlabel='UMAP_0', ylabel='UMAP_1'>,
<Axes: xlabel='UMAP_0', ylabel='UMAP_1'>], dtype=object)
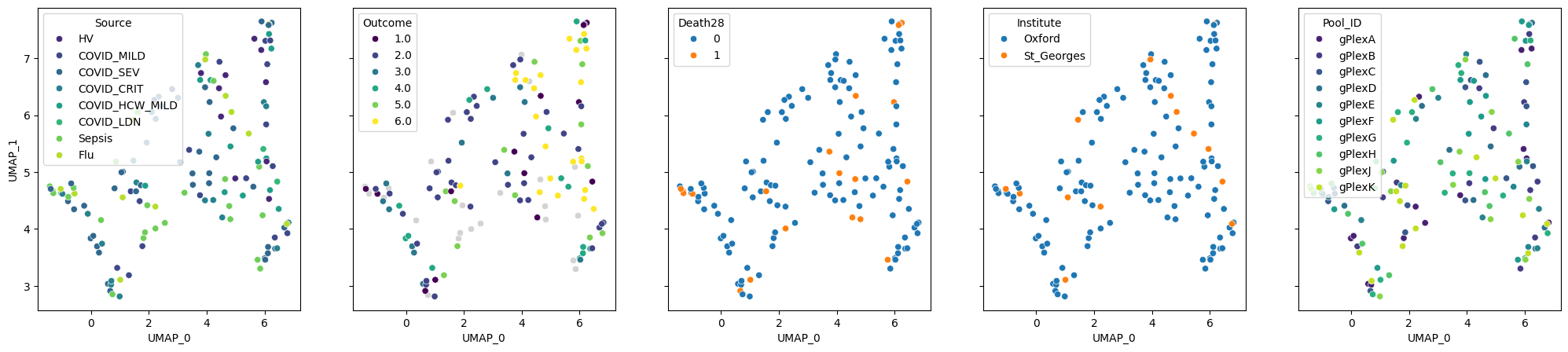
We can also visualise cell type proportions for each sample:
composition.sample_representation.plot(kind="bar", stacked=True, figsize=(10, 8), width=0.8)
plt.xticks([])
plt.legend(loc=(1.05, 0), title="Cell type");
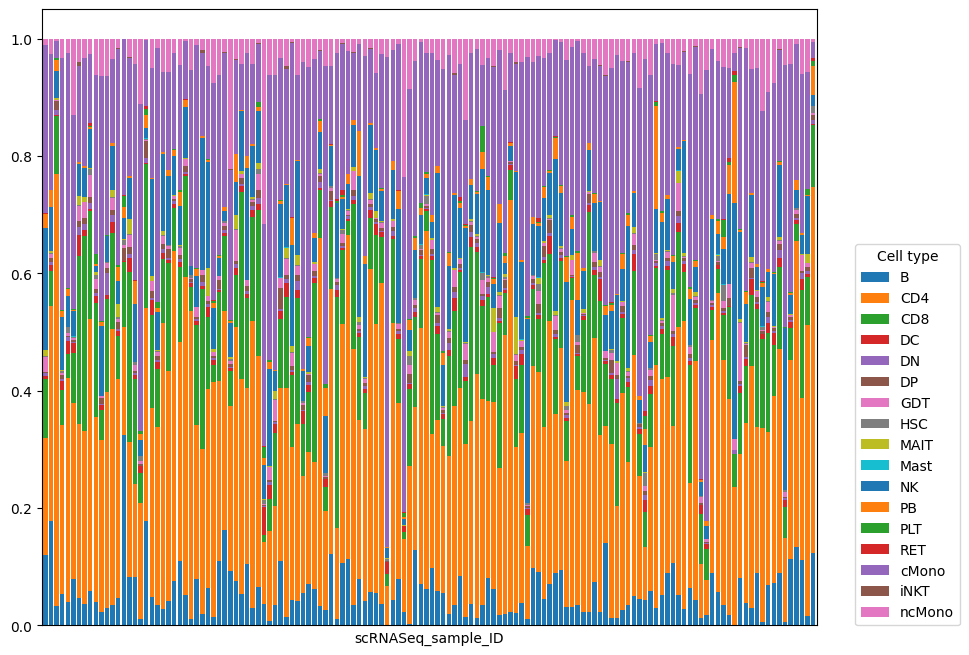
We can see that cell type composition reflects patient outcome worse than pseudobulk:
composition.evaluate_representation(target="Outcome", method="knn", n_neighbors=5, task="ranking")
{'score': 0.4739538892493282,
'metric': 'spearman_r',
'n_unique': 6,
'n_observations': 113,
'method': 'knn'}
adata.uns["composition_distances"] = composition_distances
adata.uns["composition_samples"] = composition.samples
Run PILOT representation#
PILOT is an Optimal Transport-based tool, which calculates distances between samples based on cell type proportion differences taking into account cell type similarities. Note that to run it, you need to install the dependencies additionally:
pip install patpy[pilot]
pilot = patpy.tl.PILOT(
sample_key=sample_id_col,
cell_group_key=cell_type_key,
layer="X_scVI_batch",
sample_state_col="Outcome", # It is not used for distances calculation and doesn't really matter
)
pilot.prepare_anndata(adata)
pilot.calculate_distance_matrix()
array([[0. , 0.14451829, 0.42457524, ..., 0.10953257, 0.21343671,
0.36804272],
[0.14451829, 0. , 0.32280757, ..., 0.10045083, 0.13157015,
0.25766875],
[0.42457524, 0.32280757, 0. , ..., 0.36857701, 0.23438838,
0.08670158],
...,
[0.10953257, 0.10045083, 0.36857701, ..., 0. , 0.17271665,
0.31134621],
[0.21343671, 0.13157015, 0.23438838, ..., 0.17271665, 0. ,
0.21202688],
[0.36804272, 0.25766875, 0.08670158, ..., 0.31134621, 0.21202688,
0. ]])
pilot.plot_embedding(method="UMAP", metadata_cols=samples_metadata_cols);
adata.uns["pilot_distances"] = pilot.calculate_distance_matrix()
adata.uns["pilot_samples"] = pilot.samples
adata.uns["pilot_UMAP"] = pilot.embeddings["UMAP"]
Let’s check the covariates:
pilot.evaluate_representation(target="Outcome", method="knn", n_neighbors=5, task="ranking")
{'score': 0.48835133146990367,
'metric': 'spearman_r',
'n_unique': 6,
'n_observations': 113,
'method': 'knn'}
pilot.evaluate_representation(target="Pool_ID", method="knn", n_neighbors=5, task="classification")
{'score': 0.01832203003010072,
'metric': 'f1_macro_calibrated',
'n_unique': 10,
'n_observations': 138,
'method': 'knn'}
Outcome is represented a bit worse, but Pool ID almost doesn’t affect the representation
Compare representations#
Let’s write a small function to put all the sample representations into a single object. It will make sure that the order of samples is identical and cluster the patients
def align_representations(adata, meta_adata, samples, methods):
"""
Align representations of different methods to have the same order of samples.
Additionally runs clustering with Leiden algorithm.
"""
for method in methods:
# samples_to_take = np.isin(adata.uns[f"{method}_samples"], samples)
representation_samples = adata.uns[f"{method}_samples"].tolist()
samples_order = [representation_samples.index(sample) for sample in samples if sample in representation_samples]
assert (adata.uns[f"{method}_samples"][samples_order] == samples).all(), "Order of samples is not correct"
# meta_adata.obsm["umap"] = meta_adata.obsm[f"{method}_UMAP"]
meta_adata.obsm[f"{method}_distances"] = adata.uns[f"{method}_distances"][samples_order][:, samples_order]
ep.pp.neighbors(
meta_adata, use_rep=f"{method}_distances", key_added=f"{method}_neighbors", metric="precomputed"
)
ep.tl.leiden(meta_adata, key_added=f"{method}_leiden", neighbors_key=f"{method}_neighbors")
return meta_adata
combat_methods = [
"pseudobulk",
"composition",
"pilot",
]
combat_samples = list(set(adata.uns[f"{method}_samples"]) for method in combat_methods)
combat_samples = list(set.intersection(*combat_samples))
len(combat_samples)

metadata = metadata.loc[combat_samples]
combat_meta_adata = ep.io.df_to_anndata(metadata)
combat_meta_adata = ep.pp.encode(combat_meta_adata, autodetect=True)
combat_meta_adata
! Features 'Outcome', 'Death28' were detected as categorical features stored numerically.Please verify and correct using `ep.ad.replace_feature_types` if necessary.
! Feature types were inferred and stored in adata.var[feature_type]. Please verify using `ep.ad.feature_type_overview` and adjust if necessary using `ep.ad.replace_feature_types`.
AnnData object with n_obs × n_vars = 138 × 43
obs: 'Source', 'Institute', 'Pool_ID'
var: 'feature_type', 'unencoded_var_names', 'encoding_mode'
layers: 'original'
combat_meta_adata = align_representations(
adata=adata,
meta_adata=combat_meta_adata,
samples=combat_samples,
methods=combat_methods,
)
Define types and prediction tasks for the covariates#
benchmark_schema = {
"technical": ["Institute", "Pool_ID", "n_cells", "median_QC_ngenes"],
"clinical": ["Death28", "Outcome", "Source"],
}
cols_with_tasks = {
"Institute": "classification",
"Pool_ID": "classification",
"n_cells": "regression",
"median_QC_ngenes": "regression",
"Death28": "classification",
"Outcome": "ranking",
"Source": "classification",
}
results = []
for method in combat_methods:
for covariate_type in benchmark_schema:
for col in benchmark_schema[covariate_type]:
task = cols_with_tasks[col]
try:
result = patpy.tl.evaluate_representation(
distances=combat_meta_adata.obsm[f"{method}_distances"],
target=metadata[col],
method="knn",
task=task,
# n_neighbors=5
)
except Exception as e:
print("Method:", method)
print("Col:", col)
print("Task:", task)
print(e)
print()
continue
# raise(e)
result["representation"] = method
result["covariate"] = col
result["covariate_type"] = covariate_type
if result["metric"] == "spearman_r":
result["score"] = abs(result["score"])
if covariate_type == "technical":
result["score"] = 1 - result["score"]
results.append(result)
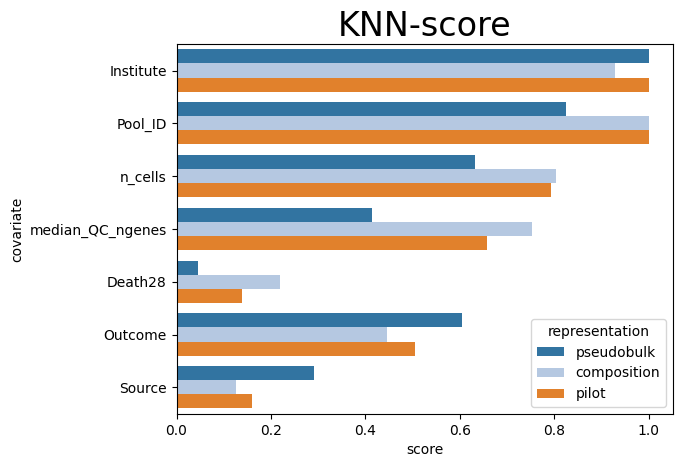
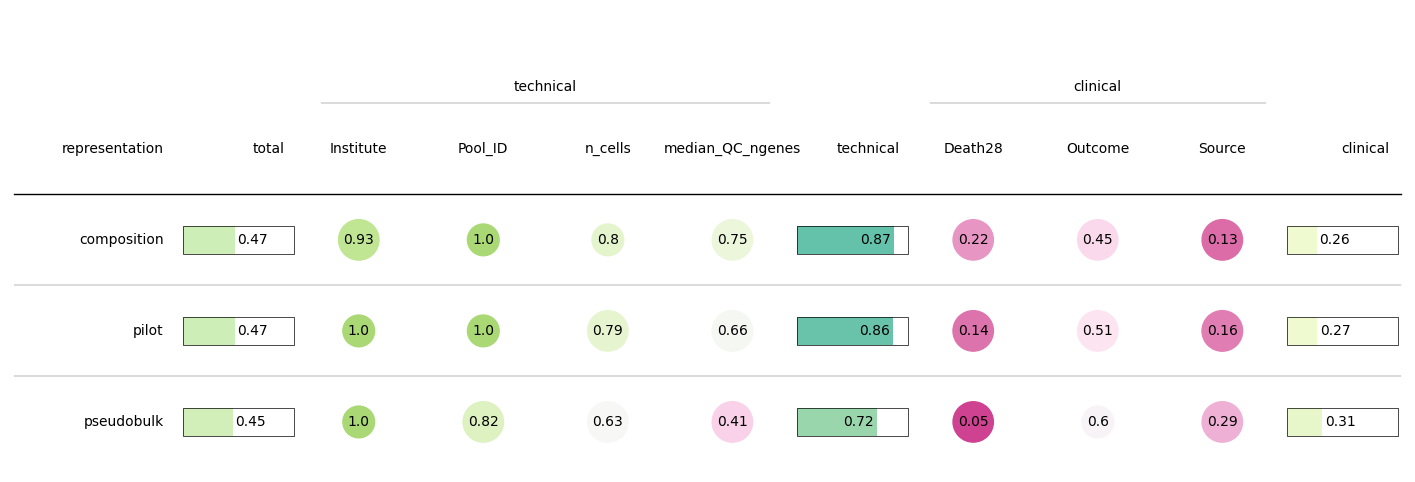
Scores are ranged from 0 to 1
For covariates that we defined as technical, 0 means that covariate strongly affects the representation, and 1 means that this covariate is randomly distributed across representation
For biological and clinical covariates, 0 means that a covariate is not represented well (it is randomly distributed), while 1 means that similar patients have similar values of covariate
knn_results = pd.DataFrame(results)
knn_results.sort_values("score", ascending=False)
| score | metric | n_unique | n_observations | method | representation | covariate | covariate_type | |
|---|---|---|---|---|---|---|---|---|
| 0 | 1.000000 | f1_macro_calibrated | 2 | 138 | knn | pseudobulk | Institute | technical |
| 8 | 1.000000 | f1_macro_calibrated | 10 | 138 | knn | composition | Pool_ID | technical |
| 15 | 1.000000 | f1_macro_calibrated | 10 | 138 | knn | pilot | Pool_ID | technical |
| 14 | 1.000000 | f1_macro_calibrated | 2 | 138 | knn | pilot | Institute | technical |
| 7 | 0.928846 | f1_macro_calibrated | 2 | 138 | knn | composition | Institute | technical |
| 1 | 0.824655 | f1_macro_calibrated | 10 | 138 | knn | pseudobulk | Pool_ID | technical |
| 9 | 0.803490 | spearman_r | 138 | 138 | knn | composition | n_cells | technical |
| 16 | 0.792614 | spearman_r | 138 | 138 | knn | pilot | n_cells | technical |
| 10 | 0.752516 | spearman_r | 119 | 138 | knn | composition | median_QC_ngenes | technical |
| 17 | 0.656404 | spearman_r | 119 | 138 | knn | pilot | median_QC_ngenes | technical |
| 2 | 0.632495 | spearman_r | 138 | 138 | knn | pseudobulk | n_cells | technical |
| 5 | 0.603334 | spearman_r | 6 | 113 | knn | pseudobulk | Outcome | clinical |
| 19 | 0.505120 | spearman_r | 6 | 113 | knn | pilot | Outcome | clinical |
| 12 | 0.446273 | spearman_r | 6 | 113 | knn | composition | Outcome | clinical |
| 3 | 0.414135 | spearman_r | 119 | 138 | knn | pseudobulk | median_QC_ngenes | technical |
| 6 | 0.292073 | f1_macro_calibrated | 8 | 138 | knn | pseudobulk | Source | clinical |
| 11 | 0.219991 | f1_macro_calibrated | 2 | 138 | knn | composition | Death28 | clinical |
| 20 | 0.160770 | f1_macro_calibrated | 8 | 138 | knn | pilot | Source | clinical |
| 18 | 0.137885 | f1_macro_calibrated | 2 | 138 | knn | pilot | Death28 | clinical |
| 13 | 0.125450 | f1_macro_calibrated | 8 | 138 | knn | composition | Source | clinical |
| 4 | 0.046083 | f1_macro_calibrated | 2 | 138 | knn | pseudobulk | Death28 | clinical |
# plt.figure(figsize=(10, 20))
sns.barplot(data=knn_results, y="covariate", x="score", orient="h", hue="representation", palette="tab20")
plt.xlim(0, 1.05)
plt.title("KNN-score", fontsize=24)
Text(0.5, 1.0, 'KNN-score')
knn_results_wide = knn_results.pivot(index="representation", columns="covariate", values="score")
# knn_results_wide.reset_index(inplace=True)
# Order the columns as in benchmark schema
cols_order = ["total"]
for covariate_type in benchmark_schema:
cols_order.extend(benchmark_schema[covariate_type])
cols_order.append(covariate_type)
cmap = LinearSegmentedColormap.from_list(
name="bugw", colors=["#FF9693", "#f2fbd2", "#c9ecb4", "#93d3ab", "#35b0ab"], N=256
)
col_defs = []
col_defs.append(
ColumnDefinition(
"total",
width=0.7,
plot_fn=bar,
plot_kw={
"cmap": cmap,
"plot_bg_bar": True,
"annotate": True,
"height": 0.5,
"lw": 0.5,
"formatter": lambda x: round(x, 2),
},
)
)
for covariate_type in benchmark_schema:
type_cols = benchmark_schema[covariate_type]
for col in type_cols:
col_def = ColumnDefinition(
name=col,
width=0.75,
formatter=lambda x: round(x, 2),
textprops={
"ha": "center",
"bbox": {"boxstyle": "circle", "pad": 0.35},
},
cmap=normed_cmap(knn_results["score"], cmap=matplotlib.cm.PiYG, num_stds=2.5),
group=covariate_type,
)
col_defs.append(col_def)
knn_results_wide[covariate_type] = knn_results_wide[type_cols].mean(axis=1)
col_defs.append(
ColumnDefinition(
covariate_type,
width=0.7,
plot_fn=bar,
plot_kw={
"cmap": cmap,
"plot_bg_bar": True,
"annotate": True,
"height": 0.5,
"lw": 0.5,
"formatter": lambda x: round(x, 2),
},
)
)
# col_defs.append(type_cols)
clin_weight = 2 / 3
knn_results_wide["total"] = (
clin_weight * knn_results_wide["clinical"] + (1 - clin_weight) * knn_results_wide["technical"]
)
fig, ax = plt.subplots(figsize=(18, 6))
Table(knn_results_wide[cols_order].sort_values("total", ascending=False), column_definitions=tuple(col_defs), ax=ax)
<plottable.table.Table at 0x42cf8fd50>
Other methods#
There are other methods implemented that you might want to try:
Aggregated per cell type (or some other group) pseudobulk:
patpy.tl.GroupedPseudobulkComposition differences:
patpy.tl.CellGroupCompositionscPoli:
patpy.tl.SCPoliMrVI:
patpy.tl.MrVIPhEMD:
patpy.tl.PhEMDMOFA:
patpy.tl.MOFAGloScope:
patpy.tl.GloScope
In the sample representation benchmark, we found GloScope to be the best representation method. For running it, R installation is required. You can use environment from this repository or manually install required dependencies.